Science postings here have been a bit light recently. I got a new job a bit back and it’s been keeping me pretty busy catching up on DNA stuff I haven’t really used since undergrad. Things are finally starting to settle down so I figure I’ll write a few posts about stuff I’ve been learning. So a lot of my job is helping to analyze the data from a shiny new DNA sequencer. Before I started, I didn’t know how far sequencing had improved in the last several years.
Until recently, most sequencing was done with Sanger sequencing. This type of sequencing produces about 100,000 bases per run and requires the DNA to be first grown in bacteria before sequencing. Then Margulies and a bunch of coauthors from a company called 454 published a paper in Nature and produced a commercial sequencer capable of sequencing 250 times as many bases per run. To do this, they used a technique called pyrosequencing. The process is pretty cool as shown in this figure from the paper.
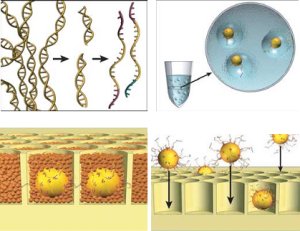
The figure goes in a clockwise direction. On the top left, DNA is fragmented into many pieces. Next in the upper right, the DNA is bound to tiny beads, one piece to a bead and the beads are isolated in little bubbles where the attached DNA is copied millions of times. This leaves each bead with millions of copies of a single piece of DNA. Importantly all these DNA are single stranded and looking for a matching strand. In the bottom right, the beads are deposited one to a well in a fiber optic slide. Then helper immobilization and enzyme beads fill in the wells in the bottom left. You can see some real images of this process in their next figure.

The left photo shows one of the beads (thin arrow) in a droplet (thick arrow). The bead is about 1/30 mm in diameter and the droplet about 1/10 of a mm. On the right is a electron micrograph of the wells on the fiber optic slide where beads are trapped. Each well is about 1/20 mm wide.
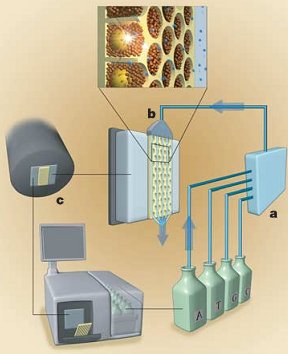
Once all this has been setup, they get to the real pyrosequencing part. With all the beads firmly nested in their separate wells, the sequencing machine takes turns flowing the A, T, C and G nucleotide building blocks of DNA over the wells. Because the DNA bound to the bead is single stranded, these new nucleotides begin building the second strand nucleotide by nucleotide. The trick to this technique (and where its name comes from) is that when a nucleotide is incorporated pyrophospate is released. This pyrophosphate is converted to ATP (a very common energy storage molecule) by enzymes on the helper beads. The ATP then fuels a bioluminescent luciferase enzyme (like in fireflies) to produce light. A 16 megapixel camera captures this light and the number of nucleotides incorporated can be estimated from the brightness. By cycling through T, A, C, and G around 40 times, the machine can count the number of bases incorporated in each step and get an average read length of about 110 bases. You can see that process in the following figure with (a) the nucleotides ready to flow over (b) the wells with their beads and produce light which is captured by (c) the camera and analyzed.
The authors were a little worried whether the shorter 110 base sequences would be useful. So they tried to sequence a bacteria, Mycoplasma genitalium. Although it’s sort of an easy target since this bacteria has a tiny 580,000 base genome, they did get an extremely thorough 40x coverage from a single run and were able to successfully assemble an accurate sequence of the genome.
The Rest of the Story
What they don’t mention in the paper is that one sequencer costs $500,000. Each run costs about $10,000 in chemicals and reagents (still cheaper than Sanger sequencing). Perhaps unsurprisingly at those prices, the 454 company responsible for this paper was later bought (for $150 million) by Roche, one of the largest pharmaceutical companies in the world.
Reminiscent of many gadgets, early adopters buying the sequencer from this paper got kind of screwed because 454 soon came out with a new improved model able to generate sequences twice as long. It looks like they’ll soon releasing an upgrade for the second model that should allow 2-4 times as many reads and again double the length (resulting in 8-16 times as many bases as this Nature paper).
The paper is a little short of pictures direct from the sequencing process so here’s a couple from a recent run. First, here’s an example of a single flow (a T nucleotide [no visible difference from other nucleotides]) showing 13 lanes of a 16 lane slide (you can divide the slide into portions to share the run [and the cost]). You might notice a pattern in some of the lanes. That’s because lanes 1-4 and 9-12 were tests to see how much DNA per bead produced the best results with the lowest concentration on the left.

And here’s a close up of a single lane during a flow (a C this time). Each bright dot signals incorporation of a C nucleotides. Brighter dots mean there were several C’s in a row.

So a very cool technology. It’s pretty amazing that an entire bacterial genome (up to about 1.5 million bases [soon to be 6 million]) can be sequenced in one shot. Unfortunately, animals including humans have genomes of 2 billion or more bases so no one will be sequencing any individuals or endangered species without a few hundred thousand dollars to burn. But a little over ten years ago, you could get published in Science for sequencing the M. genitalium and here it was used as a simple test. It’ll be interesting to see where sequencing technology stands ten years from now.
References
Margulies, M., Egholm, M., Altman, W.E., Attiya, S., Bader, J.S., Bemben, L.A., Berka, J., Braverman, M.S., Chen, Y., Chen, Z., Dewell, S.B., Du, L., Fierro, J.M., Gomes, X.V., Godwin, B.C., He, W., Helgesen, S., Ho, C.H., Irzyk, G.P., Jando, S.C., Alenquer, M.L., Jarvie, T.P., Jirage, K.B., Kim, J., Knight, J.R., Lanza, J.R., Leamon, J.H., Lefkowitz, S.M., Lei, M., Li, J., Lohman, K.L., Lu, H., Makhijani, V.B., McDade, K.E., McKenna, M.P., Myers, E.W., Nickerson, E., Nobile, J.R., Plant, R., Puc, B.P., Ronan, M.T., Roth, G.T., Sarkis, G.J., Simons, J.F., Simpson, J.W., Srinivasan, M., Tartaro, K.R., Tomasz, A., Vogt, K.A., Volkmer, G.A., Wang, S.H., Wang, Y., Weiner, M.P., Yu, P., Begley, R.F., Rothberg, J.M. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature DOI: 10.1038/nature03959